Drug labels: friend or foe? (Proceedings)
Today's drug package insert (DPI) can be a powerful ally in the selection and judicious use of a drug. The information that it provides might be categorized as either Product Description, Product Efficacy or Product Safety with some overlap among the categories.
Today's drug package insert (DPI) can be a powerful ally in the selection and judicious use of a drug. The information that it provides might be categorized as either Product Description, Product Efficacy or Product Safety with some overlap among the categories. The order presented here may not be followed on the package insert. Paramount to understanding the use of a DPI is understanding what constitutes a PI.
Definition.
The DPI is a legal document that accompanies a finished dosing form of an approved drug product. As such, it applies only to the approved product which it accompanies. The DPI for a product includes the label on the product itself as well as any and all accompanying materials (ie, inside the package. It does not include technical monographs or advertisements that might be distributed by a manufacturer. Any use of the product in hand beyond that specifically delineated in the label constitutes extra label drug use (EDLU). While EDLU is legal (as provided for by AMDUCA) and is not a means for criminal prosecution, it does not provide protection against civil prosecution. Further, the manufacturer of the approved drug can not add to the label once approved (unless it undergoes a subsequent approval process), nor can the manufacturer otherwise promote the extralabel use of the drug. The manufacturer may sponsor post-market surveillance studies that can be cited in and provide the basis for information in monographs used for promotion of the product; however, care must still be taken to not promote a use not stated on the label. Understanding the restrictions facing a drug company as it develops its label may help the understanding of what is, and what is not, provided.
Although the information on a drug label is approved by regulatory agencies, and thus is "static", the medicine surrounding the proper use of that drug is dynamic. Changes to labels that might accommodate new findings regarding the disease targeted by the drug or its use require approval by regulatory agencies, a cost the manufacturer may not want to pursue. This is probably well exemplified for antimicrobials for which resistance might emerge across time toward the target (approved) organism.
Product Description: Drug or Drug Product Name.
For the purposes of this discussion, the product refers to the finished dosing form (that is, the approved product) which includes the combined presentation of the active pharmaceutical ingredient (API; often referred to as the drug substance, which may or may not include the salt [co-ion] or ester modification), the route (which may not be included if implied by the dosing form), and the dosage form (tablet, solution, topical); release characteristics (eg. extended release tablets) may be included. Each drug label will provide three names to the product: a generic name for the drug product, which includes the generic name for the API, and the trade name for the specific drug product. The tradename can be used only by the pioneer company; thus a generic therapeutically equivalent drug product will either have no trade name or a different trade name. The generic API is named internationally and adopted nationally; the USP provides the name of the generic drug products and the manufacturer provides the trade name. Note that the manufacturer may change the API of a tradename product (more common for over the counter products).
In addition to the API, the product description will include any excipients, including fillers, stabilizers, or preservatives. These should be assessed as well for components that may contribute to allergies (ie., meat flavoring) or adversities (benzyl alcohol as preservatives for cats, xylitol as flavoring agent or carrier in dogs).
Dosage and administration:
USA dosing units for veterinary products have not been standardized to kg and thus are often offered in terms of lbs and kg. Additional very relevant, information may appear in this section, so reading through the information at least once may be prudent. Attention should be given to the basis of dosing for API modified as salts or esters: is the dose based on the total weight or the active ingredient? This becomes particularly important if moving from one dosing form of the drug to another. Newer drugs generally are named and dosed based on the active ingredient. However, older drugs may be described and dose on either/or. The best examples are the injectable chloride products, all described as 10%, but the active amount of chloride per ml varies from 0.09 to 0.4 mg/ml, or 4 fold difference. Preparation for use: This information becomes particularly important for those drugs whose strength may be impacted by inappropriate reconstitution or storage. Any variation beyond that stated on the label may increase the risk of loss of activity. One can not assume that freezing, refrigeration or other storage conditions will not negatively impact the drug. Often the information is known but can not be included on the label if it is not relevant to the approved use and indication. Storage information: As with dilution, although other storage conditions might be adequate for the product, it is only at the conditions listed on the label that stability has been proven. It should not be assumed that freezing will maintain drug stability. Products provided in protective packaging generally should be assumed to need the packaging to remain stable. The manufacturer may have additional information that could not be included on the label and may be able to share the information if specifically queried.
Expiration Date:
Generally, the expiration date should be respected although the manufacturer may have additional information and may be willing to share this if specifically queried.
Indications:
The approved indication often is not the applied indication. However, any deviation from the approved indication reflects ELDU. Efficacy and to a lesser degree, safety information can be used to support an ELDU indication.
Product Efficacy: Mechanism of action:
(MOA) Understanding the proper use of a drug begins with understanding its MOA. The FDA will require scientific support for the MOA that is claimed on the DPI. As such, a MOA may not be provided. Knowing the MOA important information to understanding the potential ELDU of the drug as well as unanticipated adversities. Further, it may become important when considering combination drug therapy (e.g., for antibiotics) or proper approaches to dosing (e.g, time versus concentration dependency). The latter (concentration dependency) might be expected for a drug that acts irreversibly, is accumulated in active for at the site of action or for other reasons, and is effective at non-detectable concentrations.
Clinical Pharmacology:
The clinical pharmacology of a drug is particularly important when trying to anticipate how changes in drug disposition that may occur in the patient (compared to the normal animal) might contribute to therapeutic failure or the advent of adverse drug events. Yet, as important as drug information may be to understanding the proper use of a drug in healthy and unhealthy animals, important pharmacokinetic information may not be provided. This may reflect the fact that kinetics were not performed because pharmacokinetics may not have been required by the FDA. Frequently the approved dose is based solely on clinical efficacy. An example is cyclosporine for which pharmacokinetics have not been provided. Even if pharmacokinetic data is provided, because data has been collected in young, healthy adult animals, the data may not easily extrapolate to the target population. In general, when applying population data to a patient, confidence intervals (95th or 99th percentile) are preferred to mean + standard deviation. A point to consider is the applicability of the breed of animal used for pre-approval studies: ie, is the Beagle an appropriate model for dogs?
General statements regarding the drug disposition (described mathematically as pharmacokinetics) can be important: General statements or terms to look for include non-linear. For example, if dosing is non-linear, increasing doses can not be expected to result in a proportional increase in drug concentrations. The direction of the lack of non-proportionality may be important. For many drugs, an increase in dose is not associated with an increase in drug concentrations (eg, selected fluorquinolones). For others, the non-proportionality may reflect saturation of drug metabolizing enzymes, which may be associated with an increased risk of adversity, particularly for a drug characterized by a narrow therapeutic window.
A. Absorption: Both rate and extent may be important. Both determine Cmax, the maximum plasma drug concentration that occurs at Tmax. The area under the curve (AUC) following oral dosing can be used to calculate oral bioavailability (the amount of drug which reaches systemic circulation; often reported as F (fraction) when compared ot AUC after IV dosing(100% bioavailability; F=1) , corrected for differences in dose. Oral bioavailability among animals may be great, which may increase the risk of therapeutic failure or toxicity. If a safe drug, variability may be countered with using higher doses. (b). The impact of food (or other factors) on absorption should be assessed in the context of the impact on Cmax (if a concentration-dependent drug). (c). If oral bioavailability is reduced, is this due to first pass metabolism (see elimination)? If yes, the risk of adversity may be increased because of differences in metabolism. Further, the use of a human drug (including generic) might accordingly be expected to be less predictable. Reviewing potential drug interactions, and specifically, the relationship between the approved drug and P-glycoprotein would be helpful. Other conditions impacting absorption should be assessed.
B. Distribution: Assessment of the volume of distribution (Vd) is more important for drugs whose efficacy is dependent on distribution to tissues that are generally hard to penetrate. A water soluble drug (generally Vd< 0.3 L/kg) is less likely to reach such compartments compared to a lipid soluble (generally Vd≥ 0.6 Lk/kg)? Although Vd does not indicate to where the drug is distributed, it dose provide an indication of extent. For antimicrobials, whether or not the drug is likely to penetrate "tough" or sanctuary tissues, or inflammatory debris, might be assessed. In general, for all drugs, a large Vd may indicate accumulation of the drug in some tissues. Accordingly, the time to steady-state and risk of adversities may increase.
• Significant binding to serum proteins may be important. If yes, binding may prolong the time to effect, the time to maximum effect, and the duration of effect, as well as the time for elimination if an adverse reaction occurs. Because only unbound drug is active, the DPI should provide information on unbound drug, which should be the basis of therapeutic decision making. For older package inserts, failure to provide the information will limit judicious used and decisions should be based on estimated the unbound fraction from protein binding information.
•.Information regarding distribution to target tissues must be considered critically, specifically in terms of how the data was collected. Tissue homogenate data can be misleading because it includes both intracellular and interstitial tissue. The former may reflect accumulation of drugs that penetrate cell membranes, but may not be impacting the microbe. As such, it may overestimate efficacy. For water soluble drugs, intracellular concentrations may be negligible despite adequate interstitial concentrations; the latter will be underestimated.
C. Elimination. The organ of clearance should be identified. If the liver, the role of active metabolites and the impact of first pass metabolism should be assessed. The implications regarding safety and drug interactions are automatically increased if the drug is cleared by hepatic or other organ metabolism prior to excretion. If the kidney is the sole organ of excretion, the risk of nephrotoxicity may be increased. Further, if the drug is characterized by a narrow therapeutic window, dose adjustment must be determined in patients with decreased renal / hepatic / cardiac function. The elimination half-life is important because it determines time to steady-state concentrations (and thus a lag time to efficacy) or the time necessary for the drug to be eliminated in the case of adversity. It may also indicate the amount of fluctuation during a dosing interval or whether or not the drug will accumulate. For drugs characterized by a narrow therapeutic window, the half-life may indicate whether or not doses or intervals be manipulated to enhance efficacy or safety. For antimicrobials, half-life may be important for time-dependent more so than concentration dependent drugs. Population data may be particularly useful in predicting population response. However, its applicability to the target population must be adequately demonstrated. For elimination data in particular, several means may be offered. Arithmetic means are the standard. Exceptions include: Harmonic mean: generally reported (along with pseudostandard deviation) for elimination half-life. This parameter is determined from a fraction (elimination rate constant; -time [0.45-hr or 0.45/hr]. Accordingly, the data is not normally distributed, and a mean can not be calculated. The harmonic mean is determined from the inverse of half-life, which is averaged and then inversed again. Geometric mean is calculated for numbers that increase non-linearly, such as occurs for MIC (serial dilutions). The log of each number is averaged and then the antilog is reported. The lack of pharmacokinetic data can be a major detriment to extralabel use, as occurs for cyclosporine (see Class B drugs, this same proceedings).
D. Microbiology: Pharmacodynamic data (MIC) should be based on an adequate sample of the target population (ie, > 100 isolates). Note that the FDA may not allow microbiologic data for organisms not included in the approved use, despite the likely use of the drug for non-approved organisms. Data provided generally includes the range, the MIC 50 (median) or MIC 90 (90th percentile). Generally the MIC will be a concentration included in standard C& S testing, ie, a two fold dilution or increase of 2. For antimicrobials, as important as the information might be to off-label use of an antimicrobial, pharmacodynamic information may not (and is unlikely to) be included for non-approved microbes. This markedly curtails the judicious yet off label use of the antimicrobial.
• The relatively susceptibility of organisms to the drug can be compared based pharmacodyanmic data. The high end of the range for each organism is the MIC 100; this might be considered the ideal target of the sample population. However, the MIC 90 is reasonable surrogate marker of what should be targeted to treat a patient for which MIC data is not available. The distance between the MIC50 or the MIC90 may reveal the level of resistance that the organisms has to the drug of interest. This relationship can be used to build a dosing regimen (dose or interval) when considered in the context of "what is achieved" in plasma (use the Lower Confidence Interval [LCI] for Cmax or, more ideally, interstitial fluid). For example, for cefovecin, comparison of the MIC 90 of Staph. intermedius (0.25 mcg/ml ) to unbound drug in canine plasma reveals concentrations to be above the MIC (T>MIC) for 12 days if using the average, or XXX days if using the LCI. An example of what is missing on the PDI is the MIC90 for S. aureus (2 mcg/ml as reported by Stegemann 2006). Average unbound drug concentrations reach that point at 2 days in the dog, but using the LCI, are not reached at all.
E. Effectiveness: Field trials. Efficacy field trials will focus on the approved indication, will reflect use as intended in target animals, and must be controlled. Controls may be, negative placebos, for which superiority must be demonstrated and which is accompanied by ethical constraints or positive controls for which non-inferiority is generally the target. Consider the relevancy of the positive control and the indicated use, recognizing the limitations put forth to the company by the FDA. For positive controls, the FDA requires that the standard to which the new drug is compared be one that has been approved for the indication being pursed. For that reason, once an indication has been approved for a drug, "follow-on" drugs may base their approval on the same indication. An example may be abscesses as an indication for the approval of an antimicrobial in cats, recognizing that antimicrobial therapy may not be indicated for its treatment. Although atopy may be a very appropriate indication for cyclosporine, it may not be the most common indication. Accordingly, the dosing regimen may not be appropriate for ELDU.
Product Safety:
Adverse reactions:
Adverse drug reactions in the medical literature generally refer to reactions that can cause harm to a patient and as such, may require a therapeutic intervention. Side effects generally are not associated with harm but may be undesirable and may be sufficient to cause the drug to not be used. For cats and cefovecin, note the statement regarding increased BUN in 10% of cefovecin treated patients when comparing pre and post treatment. Similar data has not been provided for the placebo group, limiting the ability to make an informed assessment. Foreign Marked Adverse Reaction information reflects information that may have emerged as a result of the study or use of the drug in foreign markets. Foreign post market surveillance information (which by definition may not be on a newly approved drug) may be very applicable.
Safety Information:
Three sources of safety data may be present on a package insert.
Toxicity studies:
The approval process can not detect all adversities. Toxicity data should be understood in the context in which it was collected: normal animals, several magnitudes of the dose (ie, 3,5 or 10 X) and a duration several magnitudes beyond that recommended. This does not represent the standard target population, but may, nonetheless, provide "clues" as to potential adversities.


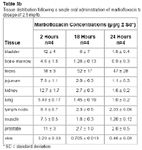
Table 3b
Field Studies:
While the sample size for toxicity studies often approximates 12, for field trials, those numbers often exceed 100 to 200 for each indication. Post-market surveillance. Despite the larger number of animals, the number may be insufficient to detect subtle adversisties. Using NSAID induced liver disease as an example (1.4/10,000 dogs for carprofen), the number of animals necessary to detect a significant difference compared to placebo for this adversity would be several tens of thousands. The incidence is even lower for fluorquinolone-induced retinal degeneration in cats, with an incidence of 1/125,000. While post-market surveillance data will not be present on the newly approved drug, it may be added post-approval if significant information emerges during drug use.



Figure 2
Note that all safety information on the label may not necessarily apply to product in hand. During the course of approval, any data generated regarding the use of the drug is reviewed by the FDA. Any information that the FDA finds relevant may be included on the label (likewise, information that the FDA finds to promote inappropriate use is likely to be excluded). For example, during the approval process for deracoxib and ferocoxib, studies were implemented using a preparation other than the approved product (decracoxib) and in animals younger than the targeted population (firocoxib; juvenile dogs). For each drug, the respective study resulted in adverse events that are included on the label but may not be relevant to the intended use. None-the-less, the information should not necessarily be ignored because it may be relevant to extra-label use. Note that a company may willingly or be compelled to change a drug label if a data emerges that suggest conditions of unsafe use. A company may also cause a label change if a new indication is approved by the FDA.
In addition to safety information, addition levels of warning to the drug user may be present on a drug label.
Contraindications represent the strongest warning and are established situations underwhich the drug should not be used. Such use should be expected to result in an adverse event. (e.g, combinations of NSAIDs, administration of a known teratogen to a pregnant animal).



Table 4
Warnings:
This may include conditions under which the drug was not tested by an adverse event might be anticipated based on the drug chemistry, action, etc.



Precautions:
Eg. for cefovecin, false glucosuria and proteinuria may be very clinically relevant.
Information for the Owner is not included for all drugs, but may emerge during post market surveillance in an attempt to reduce the risk of adversity. This information may be provided as a separate "tear" sheet that may be sent home with the client.


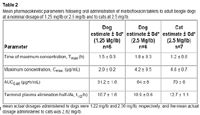
Table 2
Drug Interactions should be closely reviewed for those that put patient health at risk (ie, increased absorption, prolonged elimination. Note that drug interactions are not likely to be detected during the approval process because study subjects (toxicity or field trial) generally are receiving no drug other than the approved product. Drug interactions might be predicted based on routes of absorption or elimination, particularly as the science of pharmacogenomics advances. It is through post-market surveillance that most drug interactions are likely to emerge. As such, absence of information regarding drug interactions on the PDI should not be interpreted to imply they do not exist.



Table 4













Podcast CE: A Surgeon’s Perspective on Current Trends for the Management of Osteoarthritis, Part 1
May 17th 2024David L. Dycus, DVM, MS, CCRP, DACVS joins Adam Christman, DVM, MBA, to discuss a proactive approach to the diagnosis of osteoarthritis and the best tools for general practice.
Listen



