Sodium concentration, ECF volume, and the treatment of severe hypo/hypernatremia (Proceedings)
Sodium concentration is an expression of the relative numbers of sodium molecules to water molecules, irrespective of the total numbers.
Sodium concentration
Sodium concentration is an expression of the relative numbers of sodium molecules to water molecules, irrespective of the total numbers. Sodium content represents the total number of sodium molecules present in the extracellular fluid compartment (ECF).
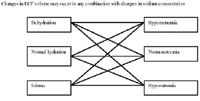
Sodium concentration is a transcellular fluid flux issue and is dealt with in this chapter. Sodium content is a volume issue heralded by physical indications of hypovolemia or dehydration/edema, and is dealt with in chapters dealing with hypovolemia and dehydration. Abnormalities in sodium concentration (hyponatremia; hypernatremia) may occur in any combination with abnormalities in sodium content (dehydration; edema). Laboratory measurements (packed cell volume, total protein concentration, and electrolyte concentrations) determine the nature of the fluid loss and the nature of the fluid replenishment. Polycythemia or hypernatremia do not define dehydration.
Free water is qualitatively expressed in terms of the sodium concentration, compared to normal for the fluid in question. Free water must be added to reduce a sodium concentration from 145 mEq/L to 120 mEq/L, for instance. Free water excess is synonymous with hyponatremia and is caused by a free water gain. Hypernatremia is a free water deficit and is caused by free water loss. Free water may be lost via evaporation (lungs and skin) or by losing fluids which are low in sodium compared to normal ECF (diarrhea, vomitus, urine). Free water may be gained by drinking water or may be administered in the form of 5% dextrose in water.
Urine sodium concentration normally varies between 60 and 100 mEq/L. Lower urine sodium values represent free water loss (which may result in a free water deficit in the ECF - hypernatremia), while higher values represent a lack of free water loss (which may result in a free water excess in the ECF - hyponatremia).
Free water losses and gains change sodium concentration with minimal changes in blood volume (e.g. free water loss will cause hypernatremia but not much hypovolemia). Sodium concentration and sodium content are different in concept and they do not overlap; concentration and content abnormalities commonly occur together. Sodium concentration abnormalities are water abnormalities. Edema/dehydration are sodium content or "saline" or "crystalloid" abnormalities.
ECF sodium content (aka ECF volume)
In contrast to sodium concentration which refers to the number of sodium molecules compared to the number of water molecules, sodium content refers to the total number of sodium molecules (and the associated anion molecules) and the water molecules in the extracellular fluid compartment. Clinical assessment of ECF sodium content is the evaluation of hydration (dehydrated, normally hydrated, edematous). Variations in sodium concentration and sodium content (ECF volume) can occur together in any combination (i.e. dehydrated patients may be hypernatremia, normonatremic, or hyponatremic). "Dehydration" is defined as the loss of a crystalloid (a fluid which contains sodium and an associated anion such as chloride or bicarbonate) from the ECF; "Edema" is defined as the gain of a crystalloid (a fluid which contains sodium and an associated anion such as chloride or bicarbonate) from the ECF. These fluids do not have to contain the same concentration of sodium or any other electrolyte as is normally present in the plasma or ECF. When an animal loses, for instance, a fluid with a relatively low sodium concentration, the fluid left behind is high in sodium (the reason why most dehydrated patients are hypernatremic). If that same animal drinks a lot of water, it may dilute the sodium concentration and may induce hyponatremia, even though they are still dehydrated. A dehydrated patient is, by definition, deficient in ECF crystalloid; an edematous patient has an excess of ECF crystalloid. Whether or not this patient is hypo/normo/hypernatremic depends upon the nature of the crystalloid loss or gain that caused the dehydration or edema.





Assessment of ECF volume
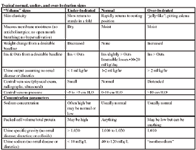
The deficit volume is usually determined by the magnitude of the decrease in skin elasticity. Normally the skin over the thorax, after being lifted into a fold, will snap immediately back to its resting position when released. When the skin fold returns a little slowly, the animal is said to be 5% of its body weight dehydrated. When the skin fold stands in the fold after it is released, the animal is said to be 12% of its body weight dehydrated. Intermediate "skin fold return rates"are extrapolated between 5 and 12 %. Unfortunately, this sign, as a quantitative estimate of the magnitude of dehydration is not very accurate; we use it because it is one of the few quantitative estimates available at presentation. Obesity will obscure the sign (obese animals may have no decrease in skin elasticity even though dehydrated); emaciation will amplify the sign (emaciated animals will have a decrease in skin elasticity even though they are not dehydrated). In addition, there is considerable patient-to-patient variation in this sign.
An acute change in body weight provides a quantitative guide to the volume of the deficit. At initial examination, however, the pre-illness body weight is seldom known. Body weight is a good way to track the adequacy of the ongoing fluid therapy plan. Lean body mass is normally neither lost nor gained rapidly enough to affect major day-to-day changes in body weight. Large volumes of fluid can, however, accumulate in cavities such as the intestinal lumen, the peritoneal or pleural cavities, or in tissues around fracture or trauma sites and effectively decrease extracellular fluid volume without a change in body weight.
Determining the magnitude of dehydration in a patient is, at best, an inaccurate science, and at worst, a wild guess. In matters of hydration status, it is always important to assess assess as many parameters as possible, and to correlate them with each other and other patient abnormalities. The end-product of this process will be to determine whether the animal is under- or over-hydrated, and, if dehydrated, to establish a quantitative estimate of the magnitude of the dehydration. If the patient is deemed to be dehydrated, the clinician may pick a number between "5 and 12% and multiply this by the animal's body weight to determine the volume of fluids predicted to remedy the dehydration. Alternately, the clinician may estimated a categorical magnitude of dehydration (mild, moderate, or severe) and then equate this with or a specific percentage (6, 9, and 12%, respectively).
There are three primary fluid compartments: intravascular, interstitial, and intracellular, and the clinical signs are different for each. An intravascular volume deficit (hypovolemia) is heralded by tachycardia and vasoconstriction; an interstitial crystalloid deficit is heralded by a decrease skin elasticity and dry mucous membranes (crystalloid losses such as vomiting, diarrhea, and diuresis) are, by definition, extracellular fluid [vascular and interstitial] losses); and an intracellular water deficit (or excess) is associated with decreased mentation and is caused by acute increases (or decreases) in extracellular sodium and osmolality. Although certain disease are associated with overlapping abnormalities in each compartment (dehydrated patients are, by definition, hypovolemic, and are commonly hypernatremic; crystalloid fluid overload would be associated with an increase in vascular and interstitial volume), there are many more diseases in which the compartmental disturbances are not linked (and may be opposite) (blood loss causes hypovolemia without an interstitial deficit (dehydration); hypoproteinemia causes hypovolemia and interstitial excess (edema). It is extremely important to evaluate an animal's blood volume status (hypovolemia/hypervolemia) separately from that of interstitial volume status (dehydration/edema) separately from intracellular volume status (hyper-, hyponatremia). Each may need to be considered separately in the fluid therapy plan. You can put them together later, if they belong together.


Typical normal, under-, and over-hydration signs
Hypernatremia
Hypernatremia may occur with hypervolemia and edema, normovolemia, or hypovolemia and dehydration depending upon the underlying disease process (Table 10-2). Hypernatremia is usually associated with the lack of sufficient water intake secondary to lack of access or lack of thirst. The most common causes include low sodium gastrointestinal losses and iatrogenic.
94% of in-hospital patients in one study (Palevsky, 1996) developed hypernatremia secondary to inadequate water intake, either due to impaired thirst or insufficient free water administration to patients with predictably increased water losses (diuretic or solute diuresis). Normal insensible losses vary with environmental and body conditions but is considered to represent about 30% of the normal ongoing losses (2-3 ml/kg/hr) or about 0.6-0.9 ml/kg/hr .
Fatal acute hypernatremia (185-190 mEq/L) was reported subsequent to drinking water provided by a faulty water softener (Hughes, 1978). Death occurred within two hours of observation of signs of ataxia, obtundation, and convulsions.
Hypernatremia causes ECF hyperosmolality and intracellular dehydration. The cells first manifesting signs of edema or dehydration are those of the central nervous system (depressed mentation, restlessness, irritability, muscle twitching/tremors, hyper-reflexivity, muscle rigidity/spasticity, ataxia, myoclonus, tonic spasms, coma). Tissue shrinkage can cause intracranial hemorrhage. In time (a day, more or less) the intracellular compartment increases its intracellular osmoles to offset the effects of the extracellular sodium aberration and to restore intracellular water volume toward normal.
First there is an accumulation of intracellular electrolytes and then there is an accumulation of organic solutes (phosphocreatinine, myo-inositols, glutamine; glutamic acid and taurine). Rapid restoration of the water imbalance at this time will cause serious water intoxication problems for the patient. Sodium concentrations should be corrected slowly; the general rule is no faster than 1 mEq/L per hour. Severe sodium aberrations may take a day or two to correct. Most sodium aberrations are, however, a secondary processes and are not severe. Effective treatment of the underlying disease process and restoration of an effective circulating volume with a neutral sodium solution will allow the animal to correct its own sodium concentration. Severe water deficits (a sodium concentration of, say, 165 mEq/L) must be treated specifically and slowly.
Volume problems should be addressed with a high sodium solution (close to but slightly below that of the patient - about 10 mEq/L below that of the plasma (this depends a great deal upon the rate of administration of the fluid). A suitable sodium concentration can be achieved by mixing saline and hypertonic saline. Access to oral water should not be permitted. The sodium concentration of the administered fluid should be gradually decreased, so as to stay about 10 mEq/L below that of the plasma. Plasma sodium concentration should be checked frequently (every 2 hours until it is at least 170 mEq/L). Furosemide could be administered as a second-order treatment to enhance urinary sodium excretion. The use of hypotonic solutions should not be used as a matter of routine, except as outlined below.



Causes of hypernatremia
The water deficit could be calculated: ({current [Na+]/normal [Na+]} - 1) x total body water (0.6 x kg of body weight). This must be administered over many hours (a few days) so as not to decrease the sodium concentration faster than 1 mEq per hour. This calculates to be about 3.7 mls of water/kg body weight/hr. Five percent dextrose in water is used as the free water source. The dextrose is metabolized and the water is distributed to all fluid compartments in proportion to their relative size. Water losses may also be replaced orally or intravenously. Sodium concentration should be rechecked often. If the sodium concentration proves resistant to this therapy in the presence of active intracranial disease, it may be that there is a lack of endogenous vasopression and exogenous vasopressin could be administered. DDAVP can be administered (1 drop into the nose or conjunctival sac every 12 hours). If expense is an issue, aqueous vasopressin could be used but it is not as effective. Chlorpropamide (enhances tubule responsiveness to vasopressin by inhibiting vasopressin stimulated production of prostaglandin E2 by medullary interstitial cells - NSAIDS have a similar effect) and carbamazepine (enhances vasopressin release) have been used in humans to treat partial diabetes insipidus.
Hyponatremia
Hyponatremia may also be associated with hypervolemia/edema or hypovolemia/dehydration (Table 10-3). Given the immense ability of the kidneys to get rid of water, hyponatremia is usually to some defect in the ability of the ascending loop and distal tubules to generate a dilute urine. The most common causes include continued water consumption in the face of crystalloid losses such as vomiting or diarrhea and impaired renal diluting capacity due to non-osmotic vasopressin secretion (dehydration; hypovolemia). Hyponatremia may also be iatrogenic.
Hyponatremia is a complication associated with head injury. The mechanisms include: 1) syndrome of inappropriate secretion of ADH; and 2) cerebral salt wasting syndrome. Non-osmotic (hypovolemic) induced ADH secretion is appropriate from the standpoint of ECF volume control but may, nevertheless, be considered inappropriate from the standpoint of promoting hyponatremia. Brain natriuretic peptide is a 32 amino acid peptide which is similar to the 28 amino acid atrial natriuretic peptide. Urodilatin is another natriuretic factor involved in the regulation of sodium and water excretion. Urodilatin is produced within the nephron by post-translational processing of a prohormone. The primary stimulus for its release are osmoreceptors in the brain and volume receptors in the heart.
In one study of subarachnoid hemorrhage (Isotani, 1994), hyponatremia was reported to be more associated with increased levels of atrial natriuretic hormone and not brain natriuretic hormone or ADH. There are small amounts of ANP in the brain, but it seems more likely that the intracranial injury somehow triggered the release of ANP from the heart. It is not clear that the central volume status was the same in normal compared to hyponatremic patients.
Dehydration/hypovolemia promote hyponatremia by non-osmotic mechanisms. Volume receptors are stimulated which then enhance the activity of the renin-angiotensin system and also activate the thirst mechanisms and vasopressin release. The decrease in renal blood flow and filtrate formation, and the increased reabsorption by the proximal tubules, decreases the delivery to filtrate to the diluting segments of the nephron, impairing free water excretion.
Hyponatremia causes intracellular edema. Hyponatremia has been associated with obtundation, anorexia, muscle weakness and wasting, and gastrointestinal signs. Common coexisting electrolyte problems include hypochloremia, hyperkalemia, and hyperphosphatemia.
Mild hyponatremia requires no special consideration beyond therapy directed to the underlying disease process and volume restoration with any ECF replacement solution. With severe hyponatremia, however, the brain compensates for the cellular edema initially by expelling some of its intracellular potassium (the primary osmotically active intracellular cation) and then the elimination of the nonionic organic solutes. Rapid correction of severe hyponatremia (<130 mEq/L) is a more serious situation and may cause myelinolysis (spastic quadriparesis, facial palsy, dysphagia, vocal dysfunction, and mental confusion to coma) (Brady, 1999; Laureno, 1997; O'Brien, 1994). Sodium concentration should not be increased faster than 0.5 mEq/L. Within hours of a too rapid correction of hyponatremia, there is an influx of electrolytes into the cells. This would normally be followed, in several days, by the intracellular increase (normalization) of the non-ionic osmoles. In the six cases reported in the veterinary literature, myelinolysis was associated with a sodium concentration increase of 0.68 to 1.0 mEq/L (Brady, 1999; Laureno, 1997; O'Brien, 1994). The associated fluid plans were: 1) 0.9% saline 22 ml/kg bolus, followed by 6 ml/kg/hr infusion (Brady, 1999); 0.9% saline at 5 ml/kg/hr (O'Brien, 1994); and 3) 3.0% saline at 4-11 ml/kg/hr for 6 hours(Laureno, 1997).



Hyponatremia
The clinical signs of myelinolysis occur 2-7 days after correction of hyponatremia. It is visible by MRI after 2 weeks. The duration is variable and recovery ranges from none to complete.
The regulation of concentrations of brain cell organic osmoles involves carrier-mediated transporters. This involves the synthesis of new transporter proteins. Rapid correction of hyponatremia results in an overshoot of brain sodium and chloride levels and this high intracellular ion concentration may underlie myelinolysis of acute correction.
Rapid, partial, correction of acute hyponatremia may be indicated to prevent brain herniation, however not as a matter of routine. Most of these patients developed their hyponatremia slowly and can be assumed to have compensated.
Volume problems should be addressed first. In severe, chronic hyponatremia, a solution that contains sodium at a concentration close to but slightly above that of the patient would be the safest. Lactated Ringers solution has a sodium concentration of 130 mEq/L and can be further diluted with 5% dextrose in water if necessary. It is potentially quite dangerous to use high sodium containing solutions such as saline or hypertonic saline unless the administration rate is monitored very closely. These fluids should be reserved for patients with hypervolemic hyponatremia. Revolumizing hypovolemic patients eliminates non-osmotic causes of vasopressin release and improve renal blood flow and thus enables such patients to more effectively eliminate the excess free water. Access to oral water should be limited or not permitted. Furosemide should be administered to enhance renal water loss. Furosemide inhibits sodium reabsorption by the ascending loop of henle which diminishes the ability of the kidney to dilute the filtrate and in turn, diminishes medullary hyperosmolality which inhibits the filtrate concentrating ability of the collecting ducts. The sodium concentration of the administered fluid should be gradually increased, so as to stay about 10 mEq/L above that of the plasma (this depends a great deal upon the rate of administration of the fluid). Plasma sodium concentration should be checked frequently (every 1-2 hours until it is at least 125 mEq/L). The tetracycline antibiotic demeclocycline and lithium have been used in humans to inhibit collecting tubule responsiveness to vasopressin.
Alternately the sodium deficit can be calculated by the formula: (measured sodium - normal sodium) x (0.6 x body weight [kg]); assuming normal [Na+ ] for dogs is 146 and cats is 156. The amount of sodium, in the form of hypertonic saline could be administered over the course of the next several days so as not to increase the sodium concentration faster than 0.5 mEq/L/hour.
The water excess required to dilute the sodium concentration to its measrued value can be calculated by the equation: ([normal sodium/measured sodium] - 1) x (0.6 x body weight [kg]).
Nonhypo-osmolar hyponatremia
Hyponatremia that is not associated with hypo-osmolality, a condition previously referred to as "false" hyponatremia, may be associated with hyperglycemia and hypermannitolemia. The increase in these solutes will draw water into the extracellular compartment which will dilute the sodium by about 2 mEq/L per 100 mg/dl increase in glucose or mannitol. These animals do not have hypo-osmolality as might be anticipated from the hyponatremia, but rather tend to be iso- or hyperosmolar (depending upon the concentration of these other solutes). This syndrome might be better name "dilutional hyponatremia?. Once the glucose is metabolized or the mannitol is excreted, the sodium concentration will return to normal.
Hyperproteinemia and hyperlipemia cause a small increase in plasma solids and volume, and a proportionate, but small, decrease in sodium concentration measured per liter of plasma when measured by the almost extinct methodology of flame photometry. This is not an issue with ion specific electrodes.
References
Brady C, Vite CH, Drobatz KJ, Severe neurologic sequelae in a dog after treatment of hypoadrenal crisis, J Am Vet Med Assoc 1999;215:222-225.
DiBartola S, Hyponatremia, in Schaer M, (ed), Veterinary Clinics of North America, WB Saunders Co, Philadelphia, 1998;28:515-532.
Fried L, Palevsky P, Hyponatremia and Hypernatremia, in Saklayen M (ed), Medical Clinics of North America, WB Saunders Co, Philadelphia, 1997;81:585-609.
Gross P, Reimann D, Neidel J, et. al., The treatment of severe hyponatremia, Kidney International, 1998;53:S 6-11.
Hughes DE, Canine Practice, 1978;5:28.
Isotani E, Suzuki R, Tomita K, et. al., Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage, Stroke, 1994;25:2198-2203.
Laureno R, Illowsky K, Myelinolysis after correction of hyponatremia, Ann Intern Med, 1997;126:57-62.
ks S, Taboada J, Hypernatremia and hypertonic syndromes, in Schaer M, (ed), Veterinary Clinics of North America, WB Saunders Co, Philadelphia, 1998;28:533-543.
Obrien D, Knoll R, Johnson G, Myelinolysis after correction of hyponatremia in two dogs, J et Intern Med 1994;8:40-48.
Palevsky P, Bhagrath R, Greenburg A, Hypernatremia in hospitalized patients, Ann Int Med, 1996;142:197-203.
Rose BD, Clinical Physiology of Acid-Base and Electrolyte Disorders, 4th ed, McGraw-Hill, Inc., San Francisco, 1994.
Zafonte R, Mann N, Cerebral salt wasting syndrome in brain injury patients: a potential cause of hyponatremia, Arch Phys Med Rehabil, 1997;78:540-542.













Podcast CE: A Surgeon’s Perspective on Current Trends for the Management of Osteoarthritis, Part 1
May 17th 2024David L. Dycus, DVM, MS, CCRP, DACVS joins Adam Christman, DVM, MBA, to discuss a proactive approach to the diagnosis of osteoarthritis and the best tools for general practice.
Listen



